Angiokeratoma
Solitary angiokeratoma; angiokeratoma of Fordyce (scrotum / vulva); angiokeratoma of Mibelli (acral); angiokeratoma circumscriptum naeviforme (segmental); angiokeratoma corporis diffusum (Fabry disease and other lysosomal storage disorders)
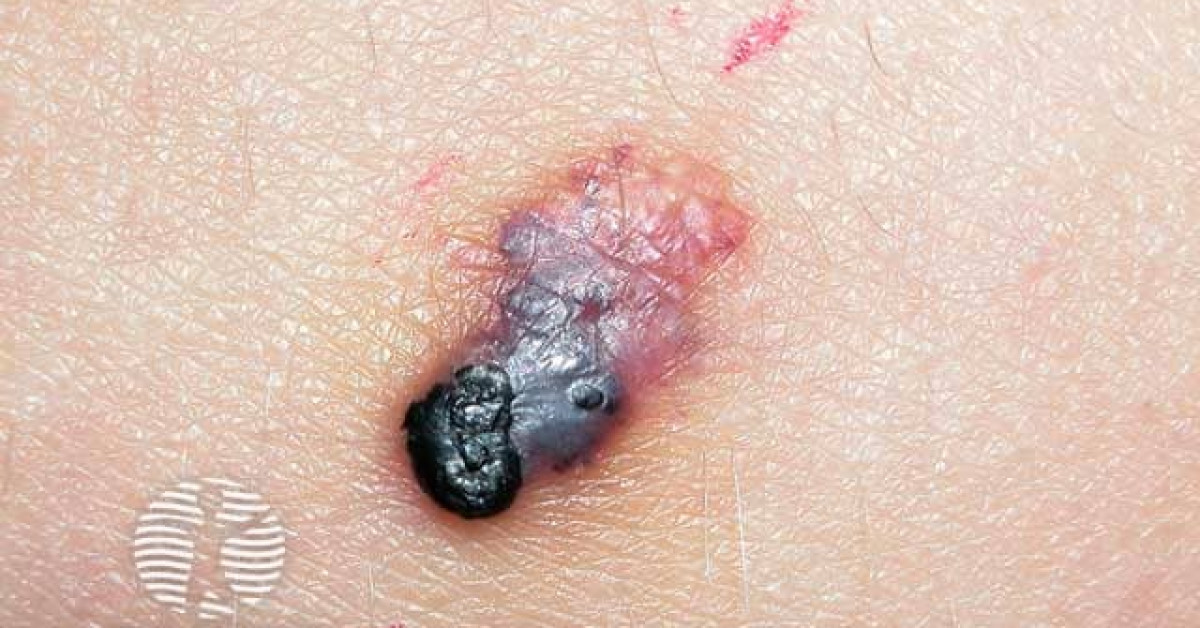
Angiokeratomas are benign vascular ectasias of the upper dermis combined with epidermal hyperkeratosis, presenting as dark red, blue or black hyperkeratotic papules. Five clinical variants are recognised: solitary, Fordyce (scrotum and vulva), Mibelli (extremities), circumscriptum (segmental), and corporis diffusum (Fabry disease and rarer lysosomal storage disorders). The skin-oncology importance is twofold: (1) the dark colour and hyperkeratotic surface make angiokeratoma one of the major clinical mimics of melanoma — particularly the solitary variant on the lower limb of an adult; and (2) angiokeratoma corporis diffusum is the cardinal cutaneous sign of Fabry disease (X-linked α-galactosidase deficiency), where prompt recognition allows life-saving enzyme replacement therapy and family screening.
Clinical variants
- Solitary / sporadic angiokeratoma — single dark red to black hyperkeratotic papule, 2–10 mm, on the lower limb / trunk; adults; the most common single-lesion variant; major melanoma mimic.
- Angiokeratoma of Fordyce — multiple 1–4 mm red-purple papules on the scrotum (men) or vulva (women); appear from middle age; benign; require no treatment unless bleeding / cosmetic concern.
- Angiokeratoma of Mibelli — multiple symmetrical dark red papules on the dorsal hands / feet, knees, elbows; adolescents and young adults; F>M; associated with chilblains and acrocyanosis.
- Angiokeratoma circumscriptum naeviforme — congenital segmental plaque on the limb; "blue rubber" appearance.
- Angiokeratoma corporis diffusum (Fabry disease) — symmetric clusters of small red-purple papules in the "swimsuit" distribution (lower abdomen, hips, buttocks, thighs, scrotum); appear in childhood / adolescence; cardinal cutaneous sign of Fabry disease.
Dermoscopy
- Dark red to red-purple lacunae arranged in clusters with overlying whitish keratotic scale.
- "Black lacunae" — sharply demarcated black-blue round structures (thrombosed vascular ectasias) — characteristic.
- Absent — pigment network, atypical vessels, blue-white veil (which would suggest melanoma).
- Caveat — solitary thrombosed angiokeratoma can be a striking melanoma mimic on dermoscopy when extensively black; biopsy any clinically uncertain lesion.
Differential diagnosis
- Melanoma — particularly nodular pigmented variant on the lower limb; biopsy any clinically uncertain solitary "angiokeratoma" of recent appearance.
- Pyogenic granuloma — fast-growing, friable, polypoid — see monograph.
- Cherry angioma — bright red rather than dark; non-keratotic — see monograph.
- Kaposi sarcoma — multiple violaceous patches / plaques in HIV / iatrogenic context — see monograph.
- Cutaneous angiosarcoma — older patient; head / scalp; bruise-like — see monograph.
- Pigmented BCC — pearly, telangiectatic, less keratotic.
Angiokeratoma corporis diffusum and Fabry disease
- X-linked deficiency of α-galactosidase A (GLA gene mutations); progressive lysosomal accumulation of globotriaosylceramide.
- Clinical features:
- Cutaneous — angiokeratoma corporis diffusum (cardinal cutaneous sign).
- Acroparaesthesias — burning / shooting pain in hands and feet from childhood.
- Hypohidrosis.
- Cornea verticillata (corneal whorls on slit-lamp).
- Renal failure (proteinuria, progressive renal impairment).
- Cardiomyopathy (left ventricular hypertrophy, arrhythmia).
- Cerebrovascular disease (early stroke).
- Other lysosomal storage disorders — fucosidosis, sialidosis, β-mannosidosis, aspartylglucosaminuria, Schindler disease — can also produce angiokeratoma corporis diffusum.
- Diagnosis — α-galactosidase enzyme assay (men); GLA sequencing (women, who are heterozygous and have variable but frequently clinically significant disease — not merely asymptomatic carriers).
- Treatment — enzyme replacement therapy (agalsidase α / β); chaperone therapy (migalastat).
- Refer urgently to clinical genetics, metabolic medicine and (in the UK) the National Lysosomal Storage Disorder Service.
Management
- Reassurance — no treatment necessary for typical asymptomatic lesions.
- Treatment options for cosmetic / bleeding concerns:
- Electrosurgery, cryotherapy.
- Pulsed dye laser (585 / 595 nm), long-pulsed Nd:YAG (1064 nm).
- Surgical excision for atypical / large lesions.
- Biopsy any atypical, large, ulcerated or solitary new "angiokeratoma" of recent onset to exclude melanoma and other vascular malignancies.
- Suspected Fabry disease — refer to clinical genetics / metabolic medicine; do not start any treatment without specialist input.
References
- Schiller PI, Itin PH. Angiokeratomas — an update. Dermatology; 1996.
- Mehta AB et al. Therapeutic goals in the treatment of Fabry disease. Genet Med; 2010.
Spot a correction?
If any clinical statement, citation or link on this page needs updating, please email admin@skinoncology.net with the page name, the proposed correction and the supporting source.